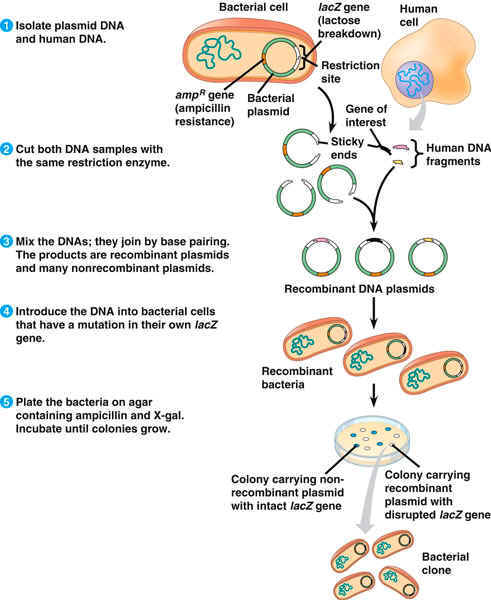
RECOMBINANT DNA TECHNOLOGY
RESTRICTION ENDONUCLEASE:
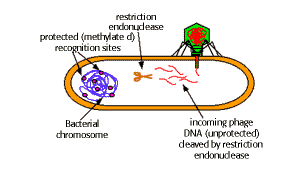
One of the most significant discoveries which allowed the development of recombinant DNA technology was restriction endonuclease. It cut double stranded DNA at specific sequences, by means of which it protects bacteria from viral infection.
The specific sequence upon which it acts may be palindromes i.e. a sequence which is the same when read in either direction. "Able was I ere I saw Elba".
There are three different classes of restriction endonucleases namely Type - I, Type - II, and Type - III.
TYPE - I:
Type - I restriction endonucleases are single multifunctional enzymes with three different subunits. They required ATP, Mg2+ and S-adenosylmethionine for its activity. Their active cleavage site usually present atleast 1000 bp away from the host specificity site. There is no enzyme turnover but translocation occurs.
Example: EcoB: TGANBTGCT
TYPE - II:
Type - II restriction endonucleases are most common type. The type - II enzymes recognize a particular target sequence in a duplex DNA molecule and break the polypeptide chains within, or near to, that sequence to give rise to discrete DNA fragments of defined length and sequence. Type - II recognition sequences are symmetric. Some sequences are continuous (e.g. GATC), some are interrupted (e.g. GANTC). Unlike type I, type II consists of a single polypeptide. The type - II require no cofactor other than magnesium ions. This type of restriction endonuclease found to be mostly used in recombination DNA technology.
Example: EcoRI : GAATTC
TYPE - III:
Type - III restriction systems are relatively rare and do not provide endonucleases for gene manipulation. Type - III endonuclease act as complexes of two subunits, one subunit (M subunit) responsible for site recognition and modification, the other (R subunit) responsible for nuclease action. DNA cleavage requires magnesium ions, ATP, and is stimulated by S-adenosylmethionine. The recognition sites are asymmetric and cleavage occurs by nicking one strand at a measured distance to one side of the recognition sequence. Two sites in opposite orientations are necessary to break the DNA duplex.
Example : EcoP1 : AGACC
If action of two different restriction endonucleases produce same cohesive ends, then the two enzymes referred as isoenzymes. For example Bam HI and Sau3AI creates the fragments with cohesive end of "GATCC" by recognizing the sequence of GGATCC and NGATCN' respectively.

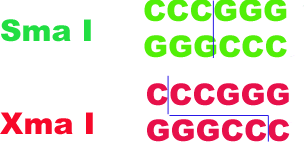
If enzymes from different sources recognize the same target , then they are said to be isoschizomers. For example: Sma I and Xma I.
NOMENCLATURE:
Nomenclature of Restriction of endonuclease followed the following points:
1. The species name of the host organism is identified by the first letter of the genus name and the first two letters of the specific epithet to form a three-letter abbreviation in italic. For example Escherichia coli --> Eco
2. Strain or type identification is written as a subscript, e.g. Ecok. In cases where the restriction and modification system is genetically specified by a virus or plasmid, the abbreviated species name of the host is given and the extrachromosomal element is identified by a subscript, e.g. EcoP, EcoR.
3. When a particular host strain has several different restriction and modification systems, these are identified by roman numerals, thus systems from Haemophilus influenza strain Rd would be HindI, HindII.,
4. Enzyme added with the prefix like endonuclease or methylase depending upon it is function. for example endonuclease R.HindIII and modification enzyme, methylase M.HindIII.
But in practice, this system of nomenclature has been simplified further:
a. Subscripts are typographically inconvenient. So, the whole abbreviation is now usually written on the line; e.g. HindIII
b. Where the context makes it clear that restriction enzymes only are involved in recombinant DNA technology, the designation endonuclease R omitted.

Restriction endonucleases allow the specific and reproducible fragmentation of DNA. The discovery of these enzymes allowed the development of modern recombinant DNA technology.
There are about 2500 different restriction enzymes which have different specificities for cutting the DNA. Sometimes the restriction enzyme cuts straight through the DNA, cutting both strands at the same location which is referred as blunt end.. Most of the time, however, restriction enzymes cut the DNA in a staggered fashion - leaving a few nucleotides of single stranded DNA extending from the cut site. These sticky ends or cohesive ends are key to allowing separate DNA molecules to get together. The short sticky ends actually can base pair between two DNA ends to align the two DNA molecules. Any two DNAs cut with the same restriction enzyme will have the same sticky ends and therefore can be joined. It is this ability provided by restriction enzymes that allows most of recombinant DNA techniques to work
Restriction enzymes, also called restriction nucleases (EcoRI in this example) , surrounds the DNA molecule at the point it seeks(sequence GAATTC). It cuts one strand of the DNA double helix at one point and the second strand at a different, complementary point (between the G and the A base). The separated pieces have single stranded "sticky-ends," which allow the complementary pieces to combine. The newly joined pieces are stabilized by DNA ligase. EcoRI, one of many restriction enzymes, is obtained from the bacteria Escherichia coli.

DNA LIGASE:
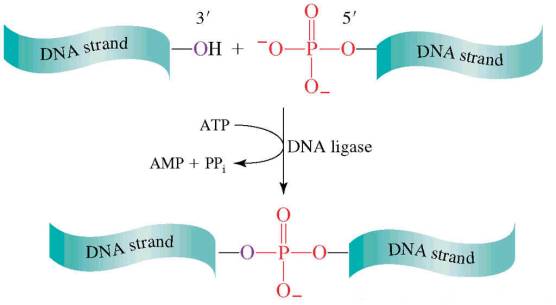
The DNA ligases are responsible for connecting DNA segments during replication, repair and recombination. They are class of enzymes that catalyze the formation of alpha-phosphodiester bond between two DNA chains. This enzyme requires the free OH group at the 3' end of other DNA strand and phosphate group at 5' end of the other. The formation of a phosphodiester bond between these groups is an endergonic reaction. Hence energy source required for ligation. In E.Coli and other bacteria NAD+ supplies the energy whereas in animals i.e. eukaryotes ATP plays the role.
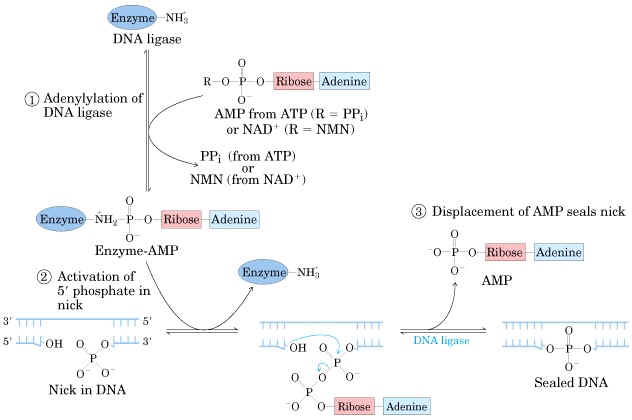
Mechanism:
a) The adenosyl group of the ATP or NAD+ is transferred to the enzyme ligase to form covalent enzyme-AMP complex in which the AMP is linked to the epsilon amino group of lysine residue of the enzyme through posphamide bond.
b) The enzyme then transfers the adenyl group to the 5' phosphoryl terminus of the nick to form a pyrophosphate grouping. In effect, AMP is attached to the 5'-phosphoryl terminus.
c) The final step is a nucleophilic attack of the 3'-OH group on the 5' activated phosphorus atom. A phosphodiester bond is formed and AMP is released. This reaction is driven by the hydrolysis of the pyrophosphate group. Thus two high-energy phosphate bonds are spent in forming a phosphodiester bridge in the DNA backbone.
VECTORS:
Vector is a DNA molecule in to which a gene is inserted to construct recombinant DNA molecule and it is also capable of replicate in a host organism. It is also otherwise known as vehicle because the vector transports the inserted gene to host organisms.

The following are the different types of vectors namely
1. plasmid
2. Bacteriophage
3.cosmid
4. Bacterial Artificial Chromosome
5. Yeast Artificial Chromosome
Sometimes vectors are referred as shuttle and expression vectors. The name "shuttle vector" represents that vector is capable of surviving and replicated in two different species or more than one organisms i.e. it might be shuttled between two different species. e.g. 2 micron circle plasmid act as shuttle vector between E.coli and yeast. Shuttle vectors allow DNAs to be transferred between two different species. The shuttle vector has two origins of replication: one that works in each host. Typically, one host is bacterial and is used for all of the cloning steps and the other host is a eukaryotic organism that can be used to study the expression from that cloned gene or can be used to synthesize a product from the gene.
Expression vector refers that the gene of interest inserted into this vector, is expressed into protein. e.g. BAC vectors usually an expression vectors. Expression vectors allow you to express certain genes directly from their recombinant DNAs. A typical expression vector will have a promoter upstream of the DNA containing the sequence to be expressed. Usually the "gene" is a cDNA because if the gene contained introns the introns would not be removed in bacteria. In addition, the promoter that is used can be an inducible one, so that synthesis of the gene product can be regulated. For example, if you wanted to express human growth hormone, you might place the growth hormone cDNA downstream of a lac promoter. In the presence of lactose, bacteria containing this construct will produce human growth hormone.
1. PLASMIDS:
It is usually a circular DNA,
Primarily independent of the host chromosome, often found in bacterial and some
other type of cells. Natural plasmids usually replicate independently of
the bacterial chromosome. plasmids are capable of having gene with upto about 10 kb size.
Many different types of plasmids have been found
in bacteria. The most useful classification of naturally occurring
plasmids is based on the main characteristic coded by the plasmid genes.
The five main types of plasmid according to this classification are as follows:
a. F plasmids:
Fertility plasmids carry only tra genes and have no characteristic beyond the ability to promote conjugative transfer of plasmids. e.g. F plasmid of E.coli
b. R plasmids:
Resistance plasmids carry genes conferring on the host bacterium resistance to one or more antibacterial agents, such as chloramphenicol, ampicillin etc., This type of plasmids mainly used in recombinant DNA technology.
c. Col Plasmids:
These plasmids code for colicins (proteins) which kill other bacterial growth e.g. ColE1 of E.Coli.
d. Degradative plasmids:
These plasmids allow the host bacterium to metabolize unusual molecules such as toluene and salicylic acid. e.g. TOL of Pseudomonas putida.
e. Virulence Plasmids:
They confer pathogenicity on the host bacterium e.g. Ti plasmids of Agrobacterium tumefaciens, which induce crown gall disease on dicotyledonous plants.
2 micron circle plasmid is an example for the plasmid which live in organism i.e. yeast other than bacteria. Two plasmid vectors that have been extensively used in genetics are pBR322 and pUC18. These vectors are derived from natural plasmids, but both have been genetically modified for convenient use as recombinant DNA vectors.
Example: pBR322
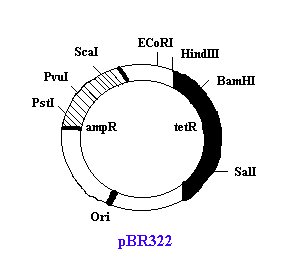
The name 'pBR322' conforms with the standard rules for vector nomenclature.
"p" indicates that this is indeed a plasmid
"BR" identifies the laboratory in which the vector was originally constructed BR stands for Bolivar and Rodriguez, the two researchers who developed pBR322.
'322' distinguishes this plasmid from others developed in the same laboratory.
It has two drug-resistance genes, tetR and ampR. Both genes contain unique restriction target sites that are useful in cloning. Gene of interest might be inserted in any one of these resistance gene and which intern is used to select the recombinant vector after transformation.
pUC plasmid:

The pUC plasmid is a more advanced vector, whose structure allows direct visual selection of colonies containing vectors with donor DNA inserts. The key element is a small part of the E.Coli beta-galactosidase gene. Polylinker or multiple cloning site which contains many unique restriction target sites used for the gene insertion, inserted into the galactosidease gene. The polylinker is in frame translationally with the galactosidase fragment and doesnot interfere with its translation. Insertion of gene of interest to this lacZ gene used for their selection in X-gal medium depending upon the color development. i.e. active enzyme expression produce blue color whereas inactive enzyme produce the white color colonies.
Selfreplicative nature and expression nature of the plasmids found to be the advantageous nature of plasmids. Low copy number, small fragment insertion size and low transformation efficiencies found to be disadvantageous nature of plasmids.
2. PHAGE VECTORS:
Viral vectors are generally are of two types namely bacteriophage vectors and single stranded phage vectors.
Bacteriophage vectors:
Thery are found to have several features which are advantageous as vectors. First lamda phage head capable of possessing 50kb length and central part of the phage genome is not required for replication or packaging of virus. So the central part can be utilized to insert gene of interest. Secondly, the recombinant vector packaged into phage head naturally. Third, the presence of recombinant vector can be directly selected by the lawn formation due to lytic mechanism. Finally, transforming effiiciency of bacteriophage found to be greater. Bacteriophage is a virus whose host is a bacterium. Bacteriophage DNA molecules are often used as cloning vectors. These vectors can take upto 20 kb size of gene of interest.
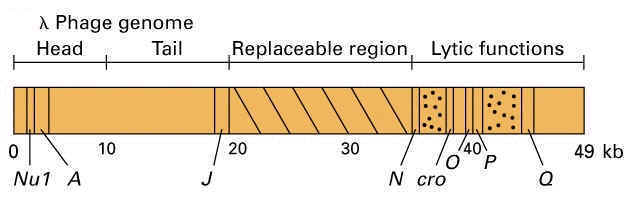
The main disadvantage of phage vector are the lysis of culture and smaller inserting fragment size.

Example: Lamda EMBL4

single stranded phage:
some phages contain only single stranded DNA molecules. On infection of bacteria, the single infecting strand is converted into a double stranded replicative form, which can be isolated and used for cloning. The main advantage is that the sequence easily sequenced through Sanger method . Example for this type of vector is M13. This virus follows lysogenic pathway so the cell lysis does not occur.
FIGURE
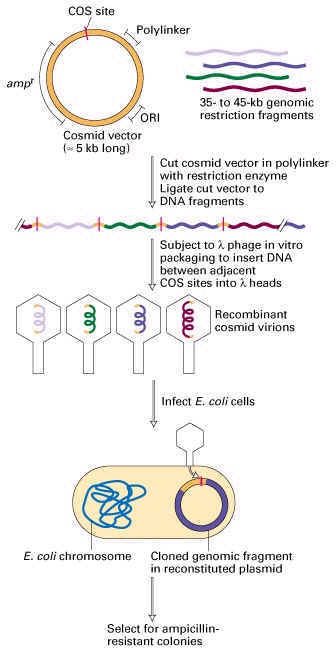
3. COSMIDS:
It is a cloning vector consisting of the phage cos site inserted into a plasmid i.e. simply plasmid and cos sites. It is used to clone DNA fragments upto 40 kb in size. cos site is one of the cohesive, single stranded extensions present at the ends of the DNA molecules of certain strains of lamda phage. The main reason for the development of the cosmid vector is to irradiate the disadvantages of plasmids and phage vectors. Due to the formation of cosmids the low copy number property of plasmids overcome because of cos sites and rolling circle replication and the lysis of culture which is the disadvantage of phage virus overcome because the vector does not have sequence for the functional phage production. After the recombinant vector multiplied, they are packaged into virus through invitro packaging method because of the cos site in phage. Transforming efficiency increased by this package method.

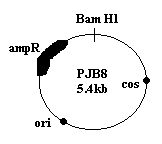
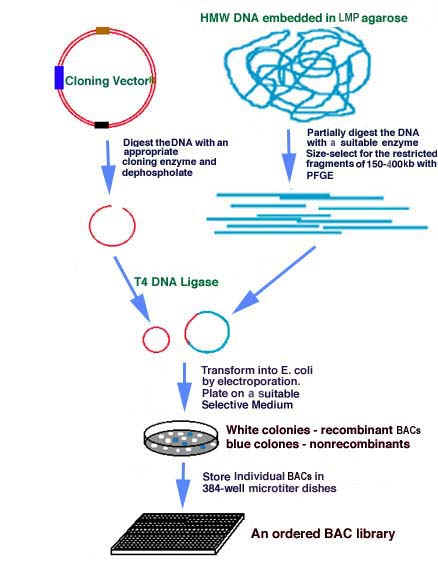
4. BACTERIAL ARTIFICIAL CHROMOSOME (BAC):
A Cloning vector based on the F Plasmid. It is used for cloning relatively large fragments of DNA in E.coli. It can handle DNA inserts upto 300 kb in size.

BAC is generally an expression vector because as indicated the figure, it contain promoter site and the vector is also significant with respect to the gene retrieval because genes contain Not I site between gene of interest.
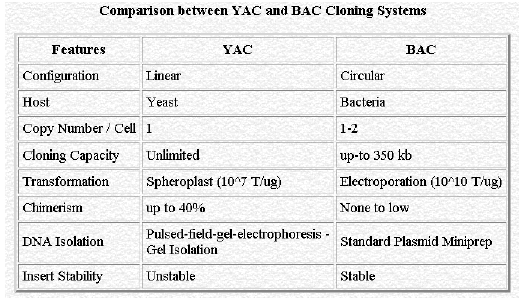
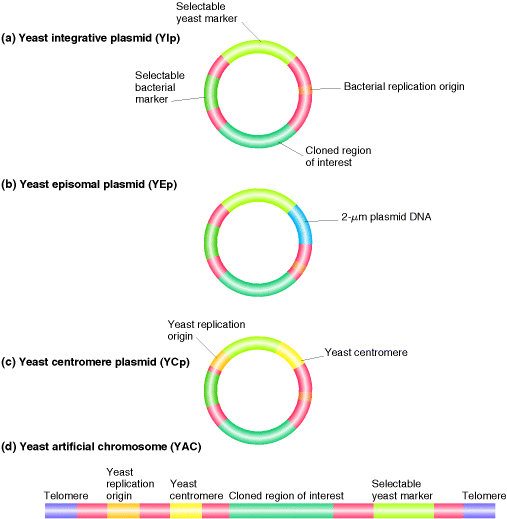
5. YEAST ARTIFICIAL CHROMOSOME (YAC):
A cloning vector comprising the structural components of a yeast chromosome and able to clone very large pieces of DNA.
Example: pYAC3
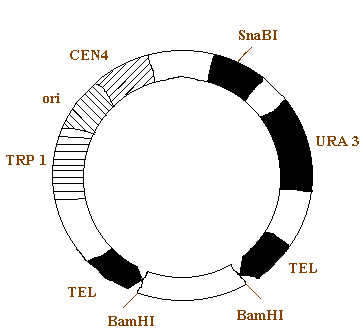
pYAC3 is essentially a pBR322 plasmid into which a number of yeast genes have been inserted. Two of these genes, TRP1 and URA 3 present in this vector which can act as selectable markers. It also contain CEN4 which is the DNA from the centromere region of chromosome 4. the TRP1- origin - CEN 4 fragment therefore contains two of the three components of the artificial chromosome.
The third component, the telomeres, are provided by the two sequences called TEL. These are not themselves complete telomere sequences, but once inside the yeast nucleus they act as seeding sequences on to which telomeres will be built. It also contain sites for the enzyme BamH1 and SnaBI. By using these two restriction enzyme the gene of interest inserted into the YAC.
The two important components of YAC, centromere and telomere have important significance. Centromere is required for the chromosome to be distributed correctly to daughter cells during cell division. Telomeres are needed in order for the ends to be replicated correctly and which also prevents the chromosome from being nibbled away by exonuclease.
YAC vectors are routinely used to clone 600 kb fragments and special types are able to handle DNA upto 1400 kb in length.

Before developing YAC, previously there different type of Yeast vectors synthesized and used. They are as follows:
PLANT VECTORS:
For plants, usually special type of plasmid known as Ti Plasmid used as vector. It is a circular plasmid of Agrobacterium tumefaciens that enables the bacterium to infect plant cells and produce a tumor (crown gall tumor)

FIGURE FROM GRIFFITH
cDNA: Human genes composed of coding and non- coding sequences. The copy of the coding sequences is called cDNA. It can be obtained from the reverse transcription of messenger RNA. The transcription and translation of the insulin cDNA will allow the production of a functional insulin molecule.
Shuttle vectors can be used to perform what has been called reverse genetics. It is possible to replace or alter the sequence of regulatory elements that control expression of a given gene then put gene back into their normal host cells to see how gene behavior has changed. This provides information on how the regulatory element might function and which are the important sequences within the regulatory element itself.
Another possibility is to put a regulatory element to be studied in front of a gene whose activity can be easily evaluated, such as the beta-galactosidase gene from the lac operon. There is a simple way to measure the level of beta-galactosidase in any cell - the enzyme turns a colorless substrate into a blue substance. Therefore, by adding this substrate to tissues, one can just measure the level of "blueness" to know the level of expression of the beta-galactosidase gene. The beta-galactosidase gene is being used as a reporter gene in this case because it "reports" on the activity of the promoter by which it is controlled. Reporter genes provide a powerful way of determining in which tissues and under which circumstances specific promoters are active.
One of the first things scientists do after isolating a specific fragment of DNA is to create a restriction map. A restriction map is simply a "map" of the locations of different restriction enzyme cut sites along the length of the DNA. Restriction maps are useful in further cloning of DNA fragments and for identifying specific regions in the cloned DNA (e.g. exons, introns, and promoters). Restriction maps can be created in sufficient detail that a restriction map can uniquely identify a piece of DNA as being different from any other piece of DNA.
STEPS IN DNA CLONING:
1. ISOLATION OF GENE OF INTEREST
2. TRANSFERRING GENE OF INTEREST INTO VECTORS
3. TRANSFERRING RECOMBINANT VECTOR INTO HOST CELLS
4. SELECTION OF THE CELLS WITH GENE OF INTEREST
5. CULTURING THE SELECTED CLONES
6. IDENTIFICATION OF PRESENCE OF GENE OF INTEREST
7. CULTURING THE IDENTIFIED CLONE FOR FURTHER GROWTH AND ISOLATION

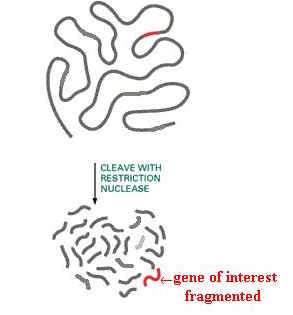
1. ISOLATION OF GENE OF INTEREST:
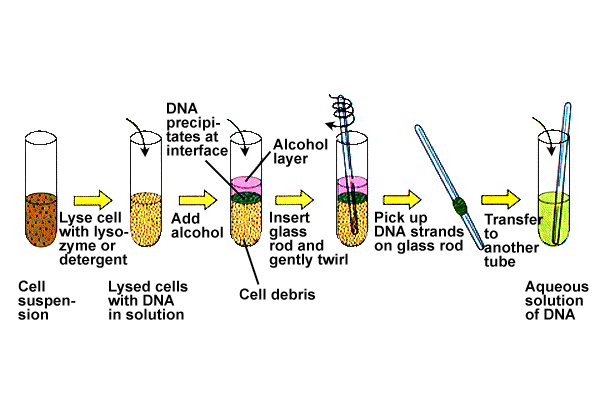
Gene of interest was first isolated. For this, initially the cells containing the gene of interest isolated and disrupted to release nucleus. From the nuclear fraction, the gene of interest released by using the restriction enzyme which posses the appropriate restriction sites at both ends of the gene of interest. After the gene of interest fragmented, they are separated by using normal isolating procedures like electrophoresis or chromatography.
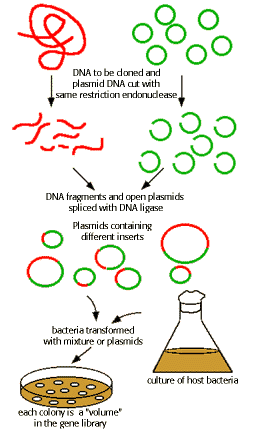
2. TRANSFERRING GENE OF INTEREST INTO VECTORS:

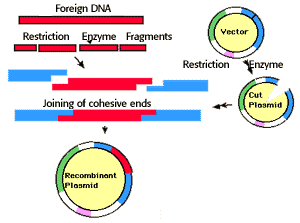
Plasmid vectors are small circular molecules of double stranded
DNA derived from natural plasmids that occur in bacterial cells. A piece of DNA
can be inserted into a plasmid if both the circular plasmid and the source of
DNA have recognition sites for the same restriction endonuclease
The plasmid and the foreign DNA are cut by this restriction endonuclease (EcoRI
in this example) producing intermediates with sticky and complementary ends.
Those two intermediates recombine by base-pairing and are linked by the action
of DNA ligase. A new plasmid containing the foreign DNA as an insert is
obtained. A few mismatches occur, producing an undesirable recombinant.
When same sticky end creating enzyme used for cleavage of vector and gene of interest, then DNA ligase seals the nick between gene of interest and vector and creates recombinant vector. Whereas when blunt end creating enzyme used then recoiling become difficult. Moreover, in both cases of using sticky end and blunt end enzymes, self coiling of vector also occur in high rate rather than the recombination of vector and genes. These situations are overcome by using
i) Linkers
ii) Adaptors
iii) Homopolymer tailing
i) Linkers:
Linker molecules are used to ligate the blunt end gene of interest with cohesive end vectors. They are normally synthesized self-complementary decameric oligonucleotiedes, which contain sites for one or more restriction endonucleases which will create sticky ends. The linker can be ligated to both ends of the foreign gene to be clones, and then treated with restriction endonuclease to produce a sticky ended fragment which can be incorporated into a vector molecule that has been cut with the same restriction endonuclease. Insertion by means of the linkers creates restriction enzyme target sites at each end of the foreign gene and so enables the foreign gene to be excised and recovered after cloning and amplification in the host bacterium.

ii) Adaptors:
When linkers added to link at the end of blunt end of gene interest, then there is an possibility of joining of multiple linkers at the end. This makes some time larger genes and waste of linker molecules. This problem is overcome by using adapters. Since adapters contain only one end suitable for joining this prevents multiple coiling of adapters. Adapter is a synthetic, double stranded oligonucleotide used to attach sticky ends to a blunt ended molecule. It contain normal 5' and 3' end at blunt end and the sticky end of adapter molecule is modified in such manner that it contain OH group on both 5' and 3' ends. This is achieved by using alkaline phosphatases. In contrast to linkers, adapters contain preformed sticky ends and joining blunt ends. Because of lack of 5' phosphate group on sticky end prevents adapter polymer formation. After the adaptors have been attached the abnormal 5'OH terminus is converted to the natural 5'P form by treatment with the enzyme polynucleotide kinase, producing sticky ended fragment that can be inserted into an appropriate vector.

Eventhough adaptors prevents polymer formation, it does not prevents self ligation or recoiling of vectors during recombination reaction. This disadvantages nature of adaptors removed by using homopolymer tailing.
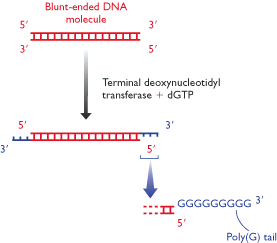
iii) Homopolymer Tailing:
A homopolymer is simply a polymer in which all the subunits are the same. Tailing involves using the enzyme terminal deoxynucleotidyl transferase, to add a series of nucleotides on to the 3'-OH termini of a double stranded DNA molecule. If this reaction is carried out in the presence of just one deoxynucleotide, then a homopolymer tail will be produced. In this method, gene of interest is tailed with one nucleotide and vector is tailed with an complementary base and when they are combined then only vector recombined with gene of interest. In this case recoiling of vectors are mostly prevented because vector does not contain complementary ends.
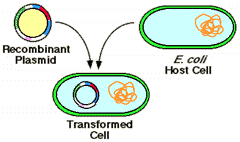
The new recombinant vector can be introduced into bacterial cells that can produce many copies of the inserted DNA . This technique is called DNA cloning.
3. TRANSFERRING RECOMBINANT VECTOR INTO HOST CELLS
The process of transferring the recombinant vector into cells usually referred by three different terminologies namely transformation , transfection and transduction. Of these word transformation commonly used.
Transformation: The introduction of any DNA molecule into any living cell.
Transfection: The introduction of purified phage DNA molecules into a bacterial cell.
Transduction: The movement of genes from a bacterial donor to a bacterial recipient with the use of a phage as the vector.
There are five different ways are available. They are as follows:
a. Precipitation
b. Protoplast fusion
c. Electroporation
d. Microinjection
e. Microprojectiles (Biolistics or gene gun)
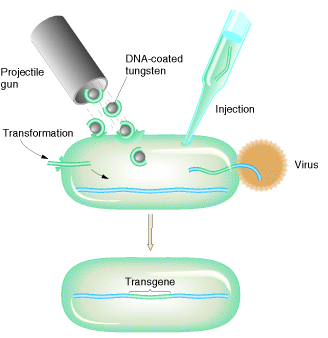
a. Precipitation:
Precipitation method involves the usage of calcium phosphate. which precipitate the DNA molecules into the surface of the host cells. Calcium plays two important role namely, first it precipitates DNA, Secondly it neutralize the charges present on the surface of cells and DNA so that they are not repelled. Once DNA in contact with the cell , they entered in to the host cells.

b. Protoplast fusion:
In plant cells usually DNA molecules transferred in to cells when they are present in protoplast form. Because in protoplast form DNA easily taken up by the cells. Protoplast is nothing but the cell without cellwall.
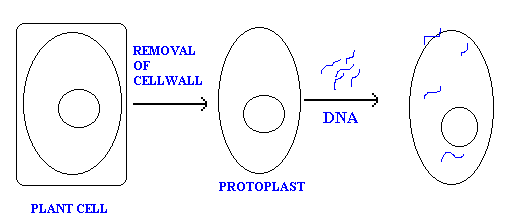
c. Electroporation:
By applying short electric pulse to the cells, a small pores were created in the cell membrane. Through which DNA can be easily transferred.
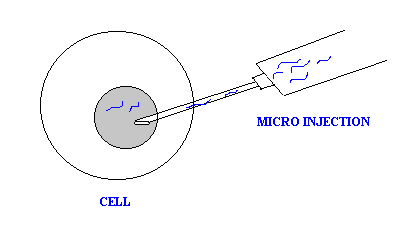
d. Microinjection:
It is a type of physical method because in this method a very fine pipette used to insert recombinant DNA directly into nucleus of the cells. This technique was initially applied to animal cells but has subsequently been successful with plant cells.
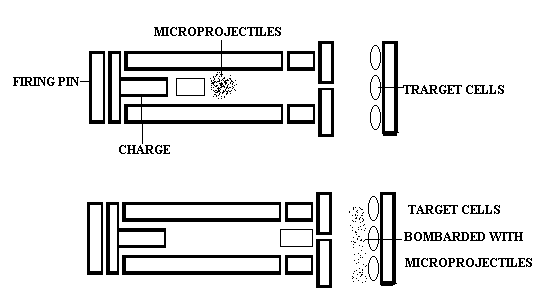
e. Microprojectiles:
The second physical method involves bombardment of the cells with high velocity microprojectiles, usually particles of gold or tungsten that have been coated with DNA. These microprojectiles are fired at the cells from a particle gun. This unusual technique is termed biolistics and has been used with a number of different types of cell.
Of the different method of transformation, precipitation, electroporation and gene gun used when the culture cells available in large number as well as where culturing of cells is not time consuming and costly. And they are usually used to transform into bacterial culture and animal cells. Microinjection usually used to transform the recombinant vector or genome into zygote or single cell where the availability and culturing condition difficult. But in the microinjection, the transformation frequency found to be maximum compared to other methods and moreover cells found to be viable. Protoplast fusion generally used to transform recombinant vector to plant cells because other methods become inefficient due to the presence of cell wall of plants.
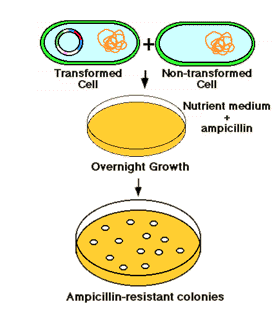
Plasmid vector contains an ampicillin resistance gene making the cell resistant to ampicillin containing medium. Growth of transformed cells (cells receiving the plasmid) can be identified on agar medium containing (e.g.) ampicillin. Thus, the cells with recombinant vector can be selected. This is an direct selection procedure.
The plasmid vector contains another identifiable gene (e.g., a second drug resistance or an enzyme activity), with the coding sequence of this gene containing the restriction site for insertion. Insertion of the foreign DNA at this site interrupts the reading frame of the gene and result in insertional mutagenesis. In the example shown below, the beta-galactosidase gene is inactivated. The substrate "X-gal" in the medium turns blue if the gene is intact, i.e. it makes active enzyme. White colonies in X-gal imply the presence of recombinant DNA in the plasmid.
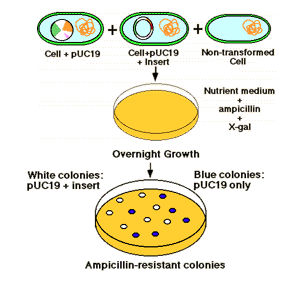
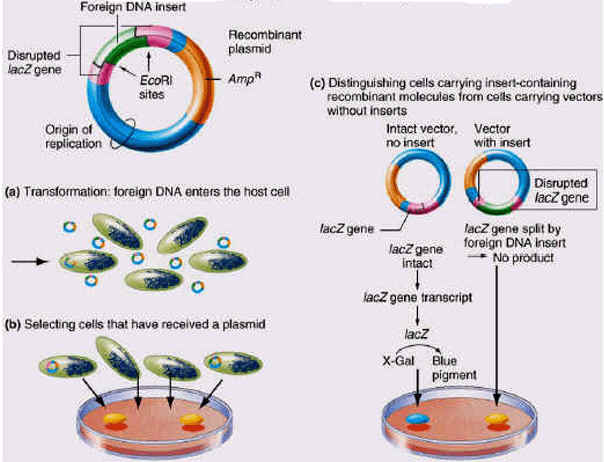
Thus by combining direct selection and insertional inactivation method used for the selection of culture with our gene of interest.
5. CULTURING THE SELECTED CLONES
Selected colonies further cultured in a separate culture plates. This makes them to culture in good nutritional medium without any limiting factor. This culture later utilized to screen (check) the presence of the perfect nature of the gene of interest and its product.

6. IDENTIFICATION OF PRESENCE OF GENE OF INTEREST
From the selected culture, the state of gene of interest screened. Screening can be carried out at the culture stage itself using replica plates. For example Insitu hybridization method. This can also be carried out using the culture medium and cell extract. For example Blotting methods. Screening carried out either by using proteins or DNA.
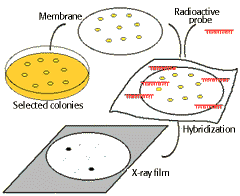
By Insitu hybridization technique:
In the following scheme, bacterial containing recombinant plasmids are grown as clones. The clones are blot transferred to a membrane sheet, and the DNA present denatured and fixed onto the surface. Adding a radioactive "probe" or complementary fragment and allowing the DNA to hybridize followed by exposure to X-ray film identifies the clone containing recombinant DNA with the correct insert.
By Southern Blotting:
DNA is taken from different colonies A and B. It is digested with restriction endonuclease. Fragments are applied onto an Agarose gel for electrophoresis. DNA has a negative charge, and in an electric field migrates towards a positive electrode. The rate of migration through a gel is proportional to the size of the fragment. DNA fragments are transferred to nitrocellulose sheets where they bind. DNA fragments are denatured and separated by gel electrophoresis. Fragments are blotted onto a sheet of nitrocellulose and fixed by heating. Blot is reacted with a radioactive probe of RNA or DNA which binds to complementary DNA. Autoradiography is used to detect radioactive fragments. The denatures fragments of DNA are fixed by baking. A radioactive probe is added. It can hybridize with a gene sequence in the DNA. The sheet is rinsed and placed next to X-ray film for autoradiography. Presence of radioactivity suggests the presence of gene of interest.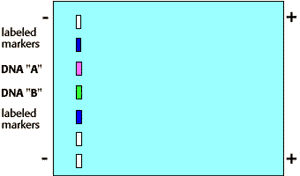

7. CULTURING THE IDENTIFIED CLONE FOR FURTHER GROWTH
After identifying the colony which was found to be containing correct gene of interest, the identified colony selected and cultured in a separate culture plates. From this culture plates gene of interest or protein was isolated and used depending upon the requirements.

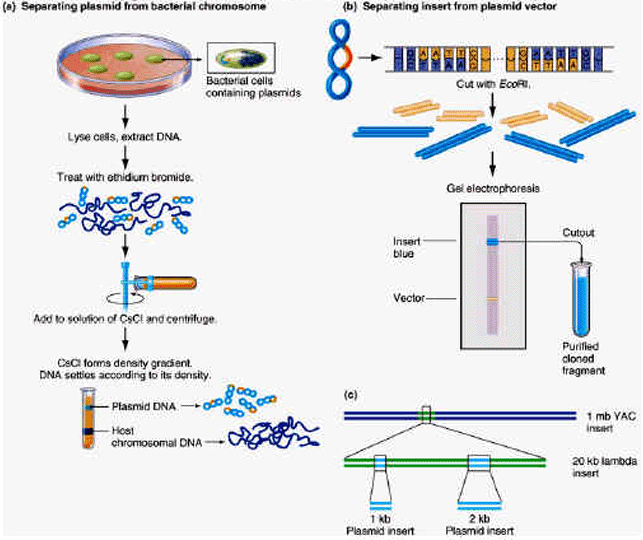
PRODUCTION OF GENOME LIBRARY:
A genomic library comprises a set of bacteria, each carrying a different small fragment of human DNA. For simplicity, cloning of just a few representative fragments (colored) is shown. In reality, all the gray DNA fragments will also be cloned.
APPLICATIONS OF RECOMBINANT TECHNOLOGY:
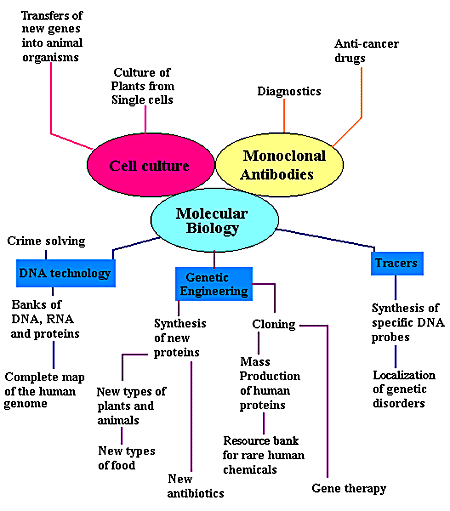
In 1885, a scientist named Roux demonstrated embryonic chick cells could be
kept alive outside an animal's body. For the next hundred years, advances in cell
tissue culture have provided fascinating glimpses into many different areas
such as biological clocks and cancer therapy. Monoclonal antibodies
are new tools to detect and localize specific biological
molecules. In principle, monoclonal antibodies can be made against any
macromolecule and used to locate, purify or even potentially destroy a molecule
as for example with anticancer drugs. Molecular biology is useful in many fields. DNA technology is
utilized in solving crimes. It also allows searchers to produce banks of DNA,
RNA and proteins, while mapping the human genome. Tracers are used to synthesize specific DNA or RNA
probes, essential to localizing sequences involved in genetic disorders.
With genetic engineering, new proteins are synthesized. They can be
introduced into plants or animal genomes, producing a new type of disease
resistant plants, capable of living in inhospitable environments (i.e.
temperature and water extremes,...). When introduced into bacteria, these
proteins have also produced new antibiotics and useful drugs.
Techniques of cloning generate large quantities of pure human proteins, which are used to treat
diseases like diabetes. .
In the future, a resource bank for rare human proteins or other molecules is a
possibility. For instance, DNA sequences which are modified to correct a
mutation, to increase the production of a specific protein or to produce a new
type of protein can be stored . This technique will be probably play a key role
in gene therapy.
Golden rice is the result of an effort to develop rice varieties that produce provitamin-A (beta-carotene) as a means of alleviating vitamin A (retinol) deficiencies in the diets of poor and disadvantaged people in developing countries. Because traditional rice varieties do not produce provitamin-A, transgenic technologies were required.
APPLICATION IN MEDICINE:
Medicine has been and will continue to be a major beneficiary of the gene cloning revolution. A number of human disorders can be traced to the absence or malfunction of a protein normally synthesized in the body. Most of these disorders can be treated by supplying the patient with the correct version of the protein, but for this to be possible the relevant protein must be available in relatively large amounts. If the defect can be corrected only by administering the human protein, then sufficient quantities will be a major problem unless donated blood can be used as the source. Animal proteins are therefore used whenever these are effective, but there are not many disorders that can be treated with animal proteins, and there is always the possibility of side effects such as an allergenic response. These proteins produced by using gene cloning. Examples for these recombinant proteins were include Insulin, growth hormone, clotting factor VIII, Interferon etc.,
PRODUCTION OF INSULIN BY RECOMBINANT TECHNOLOGY:
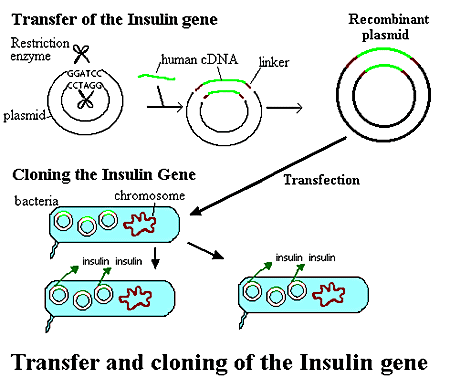
Insertion of the gene coding for insulin into a bacterial plasmid, which in turn carries the gene into a replicating bacterial cell that produces human insulin
The plasmid is cut across both strands by a restriction enzyme, leaving loose, sticky ends to which DNA can be attached. Special linking sequences are added to the human cDNA so that it will fit precisely into the loose ends of the opened plasmid DNA ring. The plasmid containing the human gene, also called a recombinant plasmid, is now ready to be inserted into another organism, such as a bacterial cell. The recombinant plasmids and the bacterial cells are mixed up. Plasmids enters the bacteria in a process called transfection. With the recombinant DNA molecule successfully inserted into the bacterial host, another property of plasmids can be exploited - their capacity to replicate. Once inside a bacterium, the plasmid containing the human cDNA can multiply to yield several dozen copies. When the bacteria divide, the plasmids are divided between the two daughter cells and the plasmids continue to reproduce. With cells dividing rapidly (every 20 minutes), a bacterium containing human cDNA (encoding for insulin, for example) will shortly produce many millions of similar cells (clones) containing the same human gene.APPLICATION IN AGRICULTURE:
Agriculture is the world's oldest biotechnology, with an unbroken history that stretches back at least 10000years. Throughout this period humans have constantly searched for improved varieties of their crop plants, varieties with better nutritional qualities, higher yields or features that aid cultivation and harvesting. During the first few millennia, crop improvements occurred in a sporadic fashion, but in recent centuries new varieties have been obtained by bredding programmes of ever increasing sophistication. However, the most sophisticated breeding programme still retains an element of chance, dependent as it is on the random merging of parental characteristics in the hybrid offspring that are produced. The development of a new variety of crop plant, displaying a precise combination of desired characteristics, is a lengthy and difficult process. Gene cloning provides a new dimension to crop breeding by enabling directed changes to be made to the genotype of a plant, circumventing the random processes inherent in conventional breeding. Two general strategies have been used:
1. Gene addition, in which cloning is used to alter the characteristics of a plant by providing it with one or more new genes.
2. Gene subtraction, in which genetic engineering techniques are used to inactivate one or more of the plant's existing genes.
A number of projects are being carried around the world aimed at exploiting the potential of gene addition and gene subtraction in crop improvement. Insect resistance , Fungal resistance, Virus resistance and Herbicide resistance plants produced by Gene addition method. Modification of flower color, reduction in starch content, Prevention of discoloration in fruits and delay of fruit spoilage were achieved with gene subtraction method.
GENE ADDITION:
|
S.NO |
GENE FOR | SOURCE OF ORGANISM | CHARACTERISTIC OBTAINED |
| 1 | Endotoxin | Bacillus thuringiensis | Insect resistance |
| 2 | Chitinase | Rice | Fungal resistance |
| 3 | virus coat proteins | Various viruses | virus resistance |
| 4 | phosphinothricin acetyl transferase | Strptomyces hygroscopicus | Herbicide resistance |
GENE SUBTRACTION:
| S.NO | TARET GENE | ENGINEERED CHARACTERISTICS |
| 1 | Polygalacturonase | Delay of fruit spoilage |
| 2 | Polyphenol oxidase | Prevention of discoloration in fruits and vegetables |
| 3 | Starch synthase | Reduction of starch content |
| 4 | Chalcone synthase | Modification of flower color |
POLYMERASE CHAIN REACTION:
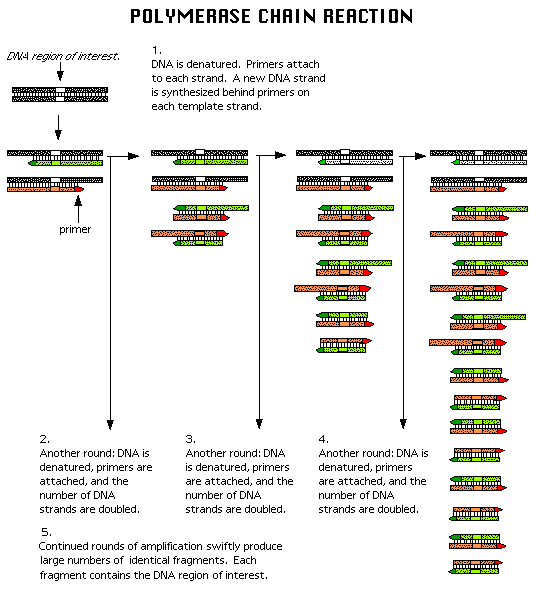
Polymerase chain reaction (PCR), is a common method of creating copies of specific fragments of DNA. The starting material is a double stranded DNA molecule. The strands are separated by heating the reaction mixture and then cooled so that the primers anneal to the two primer binding sites that flank the target region, one on each strand. Taq polymerase synthesizes new strands of DNA, complementary to the template, that extend a variable distance beyond the position of the primer binding site on the other template. The reaction mixture is heated again, the original and newly synthesized DNA strands separate. Four binding sites are now available to the primers, one on each of the two original strands and the two new DNA strands. Taq polymerase synthesizes new complementary strands, but the extension of these chains is limited precisely to the target sequence. The two newly synthesized chains thus span exactly the region specified by the primers. The process is repeated and primers anneal to the newly synthesized strands. Taq polymerase synthesizes complementary strands, producing two double stranded DNA that are identical to the target sequence. The process repeated.
Three different temperature conditions found to be maintained in PCR. They are 94*C, 30-60*C and 65 - 75*C. 94*C used for denaturation of duplex DNA into single strands. 30-60*C used to attach primers to single strands. 65-75*C used for polymerization function carried out by Taq Polymerase enzyme. PCR rapidly amplifies a single DNA molecule into many billions of molecules. In one application of the technology, small samples of DNA, such as those found in a strand of hair at a crime scene, can produce sufficient copies to carry out forensic tests.
RECOMBINANT DNA TECHNOLOGY IN BRIEF:
Many methods are available to make hybrid DNA molecules in vitro (recombinant DNA) and to characterize them. Such methods include isolating specific genes in hybrid replicons, determining their nucleotide sequences, and creating mutations at designated locations (site-directed mutagenesis). A clone is a population of organisms or molecules derived by asexual reproduction from a single ancestor. Gene cloning is the process of incorporating foreign genes into hybrid DNA replicons. Cloned genes can be expressed in appropriate host cells, and the phenotypes that they determine can be analyzed. Some key concepts underlying representative methods are summarized here.
The first step in gene cloning is to make fragments of the donor DNA by
mechanical or enzymatic methods. Certain restriction endonucleases, designated
as class II, are particularly useful for preparing defined fragments of DNA
molecules. They cleave both strands of double-stranded DNA molecules at
specific, palindromic sequences (restriction sites) that usually vary from four
to eight nucleotides in length, and the resulting DNA fragments are called
restriction fragments. Some restriction endonucleases cleave at coincident sites
to create blunt-ended DNA fragments, and others cut at staggered positions to
create DNA fragments with short, self-complementary, single-stranded 5![]() or 3
or 3![]() ends. The random probability that n adjacent nucleotides in a DNA strand will
correspond to a specific restriction site is approximately 1/4n.
Sites for enzymes that recognize unique 4, 6, or 8 nucleotide targets are likely
to occur about once in every 256, 4096, or 65,536 nucleotides, respectively. By
choosing appropriate restriction enzymes, specific DNA molecules, including
bacterial chromosomes, plasmids, and phage genomes, can be digested into sets of
restriction fragments that have appropriate sizes for specific applications.
ends. The random probability that n adjacent nucleotides in a DNA strand will
correspond to a specific restriction site is approximately 1/4n.
Sites for enzymes that recognize unique 4, 6, or 8 nucleotide targets are likely
to occur about once in every 256, 4096, or 65,536 nucleotides, respectively. By
choosing appropriate restriction enzymes, specific DNA molecules, including
bacterial chromosomes, plasmids, and phage genomes, can be digested into sets of
restriction fragments that have appropriate sizes for specific applications.
A restriction map identifies the positions of target sites for specific restriction endonucleases in a DNA molecule. Restriction maps are available for many cloned DNA fragments, plasmids and phage genomes, as well as for the entire chromosome of E coli and several other bacteria.
The second step in gene cloning is to create hybrid replicons consisting of donor DNA fragments and a cloning vector. Cloning vectors are small plasmid or phage replicons that have one or more restriction sites into which foreign DNA can be inserted. Hybrid replicons are produced by using DNA ligase to join the restricted vector DNA with donor DNA fragments that have compatible ends, or, alternatively, synthetic oligonucleotides are used as linkers to create compatibility between donor and vector DNA molecules with different ends. Ligating a vector to a heterogeneous set of DNA fragments from a donor genome is called shotgun cloning, and the collection of recombinant DNA molecules that contains the various fragments is called a genomic library. If a specific DNA fragment is available, it can be incorporated into a recombinant replicon by direct cloning into an appropriate vector chosen from the wide variety of vectors available. Plasmid and phage vectors are used mainly to clone small inserts usually less than 10 kbp. Examples of more special purpose vectors include cosmids, which are plasmid vectors that can be packaged into phage capsids (lambda cosmids accept inserts up to 30-40 kbp), and phagemids, which are plasmid-phage hybrid replicons that can exist either as plasmids or as single-stranded DNA phages under different experimental conditions. Phage P1 cosmids can accept inserts up to 100 kbp, and still larger DNA molecules can be cloned in yeast artificial chromosomes (YACs) which can stably maintain inserts up to and exceeding 1 Mbp in size. Other specialized vectors detect promoters, transcription termination signals, or other regulatory elements within foreign DNA inserts or, conversely, provide promoters from which transcription of cloned genes can be initiated.
The final steps in gene cloning are to introduce hybrid replicons into appropriate recipient cells and test them for expression of donor genes of interest. Prokaryotic cells (including bacteria) or eukaryotic cells (including yeast, animal or plant cells) can be used as recipients, but they differ with respect to their permissiveness for specific replicons, the transcriptional signals that they recognize, and the post-translational modifications of protein structure that they can accomplish. Recombinant DNA molecules produced in vitro can be introduced directly into recipient cells by transformation or transfection. In addition, clones in cosmid or phage vectors can be packaged into phage coats and introduced into susceptible recipient cells by transduction. By using specialized vectors (shuttle vectors) that can replicate in multiple cell types, genes from any organism can be cloned and manipulated in a convenient bacterial system and subsequently reintroduced into cells of the original organism for analysis in their natural environment.
Many methods are available to identify bacteria that contain recombinant DNA molecules. Most cloning vectors have genes for traits that can be positively selected, such as resistance to antibiotics. Furthermore, it is often possible to introduce foreign DNA into the cloning vector at a site that inactivates a nonessential, but easily recognizable, vector function. If both of these conditions are fulfilled, bacteria that contain recombinant molecules can be selected and distinguished easily from bacteria that contain only the vector. Bacteria in a genomic library that contain a particular cloned gene can be identified by using biochemical or immunologic methods to test for the desired gene product. Alternatively, the cloned gene of interest can be detected directly by using nucleic acid hybridization methods, provided that a specific DNA or RNA probe is available. Because insertion of foreign DNA into a cloning vector at an appropriate site does not inactivate its ability to replicate in appropriate recipient cells, hybrid replicons of interest can be amplified by replication, and the recombinant DNA molecules or their gene products can be purified and studied. The ability to purify specific DNA molecules made it feasible to develop enzymatic and chemical methods for determining their nucleotide sequences, and current methods for introducing mutations at defined sites in cloned genes are based on knowing their restriction maps or nucleotide sequences.
Recombinant DNA methods make it feasible to clone specific DNA fragments from any source into vectors that can be studied in well-characterized bacteria, in eukaryotic cells, or in vitro. Applications of DNA cloning are expanding rapidly in all fields of biology and medicine. In medical genetics such applications range from the prenatal diagnosis of inherited human diseases to the characterization of oncogenes and their roles in carcinogenesis. Pharmaceutical applications include large-scale production from cloned human genes of biologic products with therapeutic value, such as polypeptide hormones, interleukins, and enzymes. Applications in public health and laboratory medicine include development of vaccines to prevent specific infections and probes to diagnose specific infections by nucleic acid hybridization or polymerase chain reaction (PCR). The latter process uses oligonucleotide primers and DNA polymerase to amplify specific target DNA sequences during multiple cycles of synthesis in vitro, making it possible to detect rare target DNA sequences in clinical specimens with great sensitivity
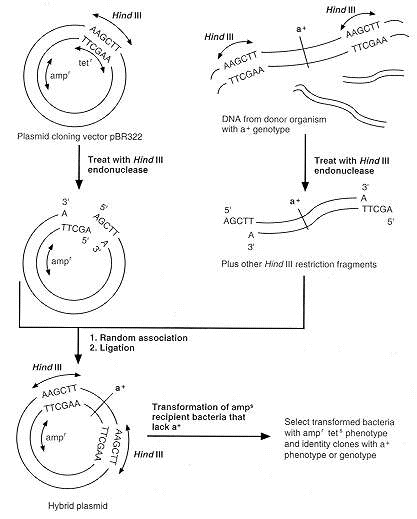
FIGURE: Plasmid cloning vector pBR322 is 4.36 kilobase pairs in size, has genes for resistance to ampicillin (ampr) and to tetracycline (tetr), and has only one HindIII restriction site that is located within the tetr locus. HindIII is used to treat samples of DNA from plasmid pBR322 and from a donor organism with a gene, designated a+, to be cloned. The donor can be a prokaryotic or a eukaryotic organism. If HindIII restriction sites are located adjacent to a+ in donor DNA, but do not occur within a+, a restriction fragment carrying intact a+ marker can be generated from donor DNA. Hybrid plasmids can be formed by random association and ligation of the HindIII-treated donor and vector DNA fragments. Although pBR322 is tetr, hybrid plasmids will be tets because the donor DNA fragments are inserted at the HindIII restriction site within the tetr locus. After transformation of amps-recipient bacteria that also lack a+, transconjugants with hybrid plasmids can be selected by their ampr tets phenotypes. Strains in which the a+ gene is present can then be identified by expresion of a+ or by testing for the polynucleotide sequence corresponding to a+. The pBR322 plasmid contains other uniuqe restriction sites that can also be used for cloning (e.g., PstI in ampr and BamHI in tetr). Many other cloning vectors and restriction endonucleases have also been used for gene cloning experiments.
APPLICATION OF RECOMBINANT TECHNOLOGY:
It can be studied in the following different headings :
1. Medicine:
2. Agriculture
3. Horticulture:
4.Floriculture:
5. Genetically engineered Microbes:
1. MEDICINE
In medicine field recombinant technology plays an vital role in prevention, diagnosis and treatment.
1.1 PREVENTION:
In prevention recombinant technology applied in two ways by vaccine production and genetic counseling
1.1.1 VACCINE PRODUCTION:
Mainly there are two examples are given under this category namely HbsAg vaccine production and DNA vaccine.
HbsAg is the surface antigen produced by Hepatitis B virus when it is infect the host cell. Host respond to this antigen and produce antibody against this antigen. By utilizing this factor when this antigen introduced into the individuals artificially they are immunized actively by producing antibodies against HbsAg. Therefore when virus infects immunized individual it is cleared off immediately by secondary immune response. HbsAg produced through recombinant technology.
FIGURE FROM KUBY
When antigen given in protein form, it might be lost during normal detoxification process but when the protein coding gene inserted to individual then it might produce the antigen for long time. So the individual found to be immune for the disease more effective and for long time. Because of these reasons gene for the antigens are prepared in recombinant vector form and injected as such usually to muscle as vaccine and referred as DNA vaccine.
FIGURE FROM KUBY
1.1.2. GENETIC COUNSELING:
This is presently followed seriously all over the world. This is nothing but comparing the genome of male and female who are going to be married and find out the possibilities of inherited disorders. Comparison of genomes carried out with the help of recombinant technology i.e. for sequencing genome, libraries should be prepared.
FIGURE FROM GRIFITH
1.2 DIAGNOSIS:
By using proteins and probes produced by recombinant technology, several diseases and disease causing agents are diagnosed. For example Malaria and AIDS are diagnosed using DNA probe and antibodies which are produced through recombinant DNA technology. Malaria diagnosed by utilizing the factor that it contains repetitive DNA sequence. Probe synthesized against this repetitive sequence. When DNA isolated from the patient serum, denatured and hybridized with the radiolabelled DNA probe, presence of malarial parasite identified through autoradiography.
AIDS identified by using antibodies against coat protein of HIV which is produced by recombinant DNA technology through western Blotting with the help of using monoclonal antibodies against HIV proteins through recombinant DNA technology.
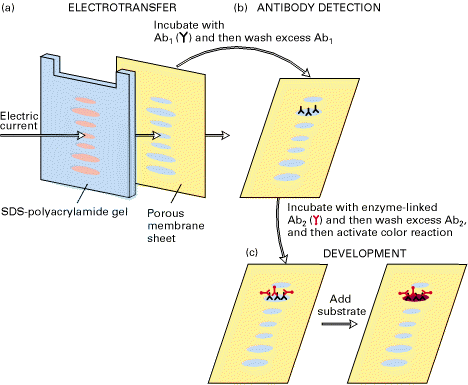
1.3 TREATMENT:
Treatment carried out by utilizing the protein product produced through recombinant technology and inherited disorders are corrected by the gene therapy.
For example, Insulin Dependent Diabetic patients are treated by insulin injection which was produced through recombinant DNA technology. Similarly Growth Hormone produced through recombinant DNA technology used to treat dwarfism. The therapeutic protein might be produced in milk. Such milk known as medicinal milk.
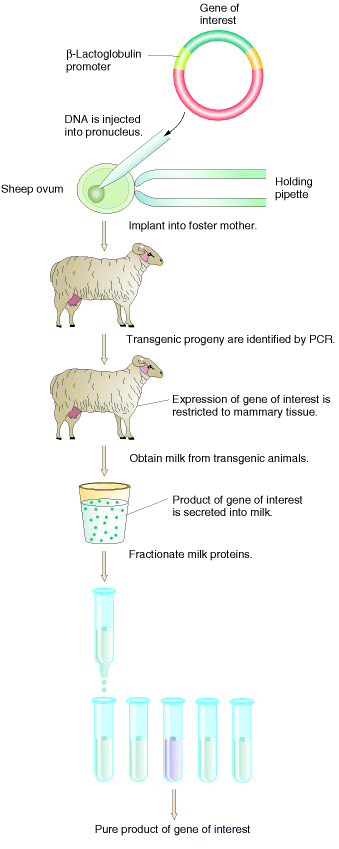
In gene therapy, the defective or absent gene given to correct the inherited disorders. For example Phenylketonuria disorder corrected by providing the gene for phenylalanine hydoroxylase enzyme. In this method genes are incorporated in vector to form recombinant vector and then this is transformed through vectors like viruses or liposomes or nanotechnology. When it reaches target cell the gene inserted into the genome and corrects the disorder. Gene therapy are of two types namely somatic gene and germinal gene therapy. Somatic gene therapy usually used to correct the disorders occuring in particular tissues only for example Diabetes Mellitus whereas germinal gene therapy used to treat the defect present in all cells of an individual for example Albinism.

2. FORENSIC SCIENCE:
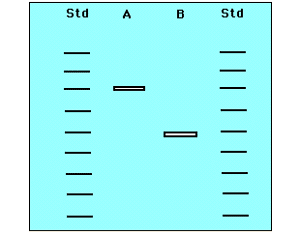
In forensic science recombinant DNA technology used to detect murderers, rapist, parents etc. There are two techniques generally used. They are DNA fingerprinting and Autoantibodies fingerprinting. In DNA fingerprinting occurrence of repetitive sequence specificity used to detect the individuals. In this technique, DNA from the suspect isolated and acted by restriction endonuclease. Then separated through electrophoresis and denatured. Then radiolabelled DNA probe against certain satellite DNA added and the number and location of occurrence specific to individual and thus it is used for detection.
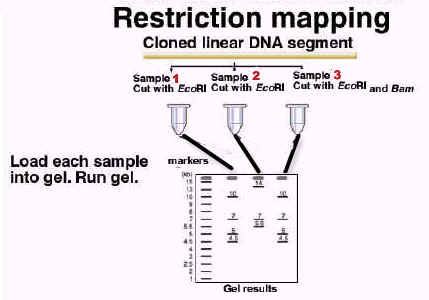
From the result it was identified that the culprit might be sample 3 because sample 1 resembles like sample 3 not sample 2.
In autoantibodies fingerprinting, radiolabelled antibodies against certain proteins which are found to be specific to individuality or varies from one to another individual used. The presence of specific proteins from the suspect and sample collected from the site of problem suggests the involvement of suspect.
3. AGRICULTURE:
In agriculture recombinant DNA technology used mainly in the following fields namely production, resistance, and nutritional improvement.
3.1 PRODUCTION:
By inducing the expression of genes in plants , the production or yiels of plants are increased. For this many inducer genes and activator genes from various species are transformed by using recombinant DNA technology.
3.2 RESISTANCE:
Resistance variety produced against different condition like herbicide, insecticide and insects. For example glyphosate is an herbicide which is generally used to remove herbs from the agricultural lands. But sometime this herbicide inhibit the growth of agricultural plants also. This is avoided by using recombinant DNA technology by inducing overproduction of the enzyme Enol pyruvyl shikimate phosphate synthase which is usually inhibited by glyphosate. Over expression achieved by using the incorporation of inducer of aro A gene from bacteria Salmonella typhimurium. Glufosinate ammonium tolerance plants are produced in the following manner:
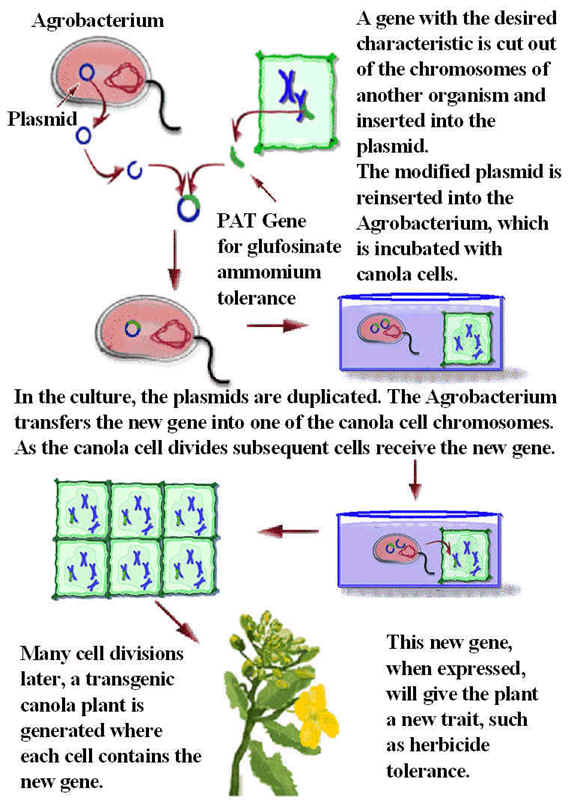
Insect resistance plants are also produced with the help of recombinant DNA technology. For example insect Manducta sexta infects various plants like tobacco, papia etc., This infection prevented by using transformation of Bt toxin gene isolated from the Bacillus thuringiensis . This gene usually produce delta- endotoxin which affects the normal digestion and absorption of insects which infects insect resistant plants which are produced through recombinant technology.

3.3 NUTRITIONAL IMPROVEMENTS:
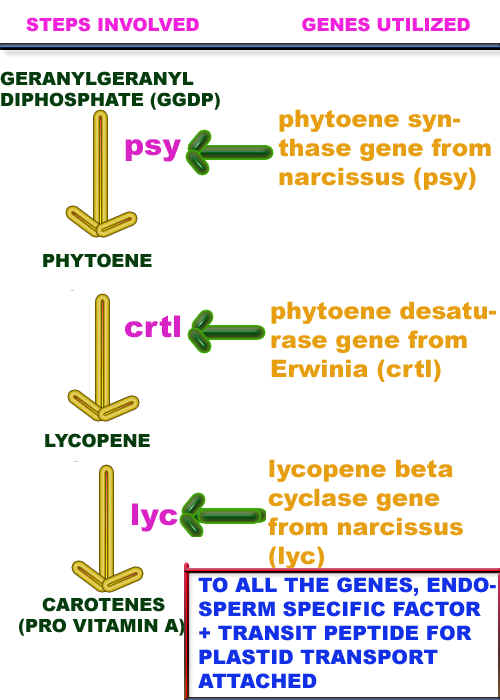
Nutritional quality of plant products improved using recombinant DNA technology. For example, provitamin A made to be produced in Rice ('GOLDEN RICE') and production of PUFA's in oil seeds.
In order to produce provitamin A in rice, the following genes are transformed to rice through recombinant DNA technology: phytoene synthase, phytoene desaturase and lycopene cyclase. Because these enzymes paly vital role in the production of provitamin A (carotenes). These genes products are also directed towards plastids so that the isolation process also found to easier.
4. HORTICULTURE:
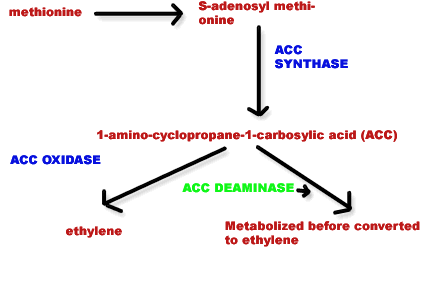
In horticulture recombinant DNA technology utilized mainly to avoid ripening. For example tomato ripening delayed by using antisense RNA against Polygalactouronase, ACC synthase and ACC oxidase since these enzymes participate in the ripening process mainly degradation of Pectin layer in the case of polygalactouronase and production of ethylene in other cases respectively.
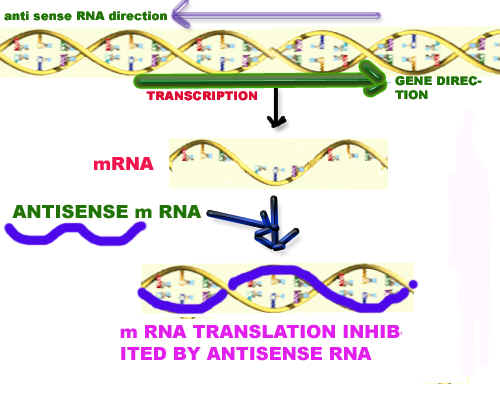
Ripening also prevented by transforming another gene known as ACC deaminase which utilizes ACC (amino cyclopropane carboxylic acid), so that it is not available for ethylene production.
5. FLORICULTURE:
Floriculture plays vital role in improving the qualities of ornamental plants. This is done by increasing the size, lifetime of flower, producing different colors and fragrance flowers.
The size of the plants are increased by simply transforming the growth hormone genes or inducing the production of growth hormone by recombinant DNA technology. Anthocyanidins are found to be responsible for the color of the flower. Depending upon the type of Anthocyanidins, flower gains the color. Thus by transforming the enzyme set up to produce specific Anthocyanidins, flower with desired color can be produced. Like preventing the ripening of fruits or certain vegetables, the life time of flower also increased using antisense RNA. Monoterpenes like geranoil , menthol found to be responsible for the fragrance of flowers. Thus by transforming genes which can produced these monoterpenes in flowers induce the fragrance in flowers.
6. GENETICALLY ENGINEERED MICROBES:
Generally microbes engineered to increase production of important biomolecules, bioremidiation and food industry. For example, biotransformation of recombinant growth hormone yield more milk in cow which is produced by genetically engineered microbes.
In bioremidiation microbes used to remove pollutants like Polyaromatic Hydrocarbons (PAH), Oil spills and heavy metals etc., Usually Pseudomonas spp. found to be used in general. Pseudomonas fluorescence isolated from PAH contaminated soils was genetically engineered with lux gene from vibrio fischeri in such a manner that in the presence of PAHs, they will emit light , which can be detected using light sensing device, thus permitting detection of PAHs in the soil. Pseudomonas capacia found to be used to degrade trichlorphenoxyacetic acid present in oil spills.
In food industry, by producing protein with all essential amino acids using recombinant DNA technology in microbes made them to utilize as food directly. For example Single cell protein concept developed by this method i.e. genetically engineered algae which capable of producing protein with all essential amino acids can be used as Single Cell Protein.